
The current COVID-1 9 pandemic is rife with questions that hackers have attacked with gusto. From 3D engraved face shields and homebrew face concealments to replacements for full-fledged mechanical ventilators, the flow of projects has been inspirational and heartwarming. At the same time there have been many efforts in a different area: investigate aimed at fighting the virus itself.
Getting to the root of the problem seems to have the most potential for close this pandemic and getting ahead of future ones, and that’s the “know your enemy” trouble that the assigned computing campaign known as Folding @Home aims to address. Millions of beings have signed up to donate cycles/seconds from spare PCs and GPUs, and in the process have created the largest supercomputer in history.
But what exactly are all these exaFLOPS being used for? Why is protein folding something to address so much computational might toward? What’s the biochemistry behind this, and why do proteins need to fold in the first place? Here’s a brief look at protein folding: what it is, how it happens, and why it’s important.
First Things First: What Do Proteins Do?
Proteins are crucial to life. They afford not only the structural elements of the cell, but too serve as the enzymes that catalyze just about every biochemical reaction. Proteins, whether structural or enzymatic, are long chains of amino acids that are related end-to-end in a specific sequence. The functions of proteins are determined by which amino battery-acids are present at various places on and in the protein. If a protein needs to bind to a positively charged molecule, for example, the binding website might be full of negatively charged amino acids.
To understand how proteins achieve the structure that defines their function, a rapid review of the basics of molecular biology and the flow of information in the cell is in order.
The production, or construction, of a protein begins with the process of transcription. During transcription, the double-stranded DNA that holds the genetic info in a cadre is partially unwound, exposing the nitrogenous groundworks of the DNA to an enzyme called RNA polymerase, often referred to as RNAPol. RNAPol’s errand is to make an RNA copy, or transcript, of the gene. This imitate of the gene, announced messenger RNA or mRNA, is a single-stranded molecule that is perfect for directing the protein manufacturing system of the cadre, the ribosomes, in a process announced translation.
Ribosomes act like a jig, taking the mRNA template and according it up to other small bits of RNA called delivery RNA, or tRNA. Each tRNA has two major active fields — one that has a three-base section called an anticodon that matches up with complementary codons on the mRNA, and a region for tying an amino acid that’s specific for that codon. During translation, tRNA molecules randomly try to bind to the mRNA in the ribosome working their anticodon. When a parallel is reached, the tRNA molecule affixes its amino battery-acid to the previous amino battery-acid, wording another link in the order of amino acids coded for by the mRNA.
This sequence of amino acids is the first tier of structural hierarchy in a protein, and is referred to as the protein’s primary organize. The part three-dimensional structure of the protein, and undoubtedly its function, comes directly from the primary organization through the different qualities of each of those amino battery-acids and how they interact with each other. If it weren’t for these chemical properties and interactions between amino acids, polypeptides would just remain linear cycles with no three-dimensional structure. We see this all the time in cooking, which is the heat-induced denaturation of the three-dimensional structure of proteins.
Long-Distance Attachments Between Duties of Proteins
The level of arrangement beyond the primary formation is cleverly called the secondary structure, and includes moderately short-range hydrogen bails between amino battery-acids. These stabilizing interactions anatomy two major motifs: the alpha-helix and the beta-pleated sheet. The alpha-helix species a tightly coiled polypeptide neighborhood, while the beta-sheet is a flat, broad-spectrum orbit. Both motifs have structural qualities as well as functional dimensions, depending on the characteristics of the amino acids within them. For sample, if an alpha-helix has primarily hydrophilic amino battery-acids within it, like arginine and lysine, it’s likely to be involved in aqueous reactions.
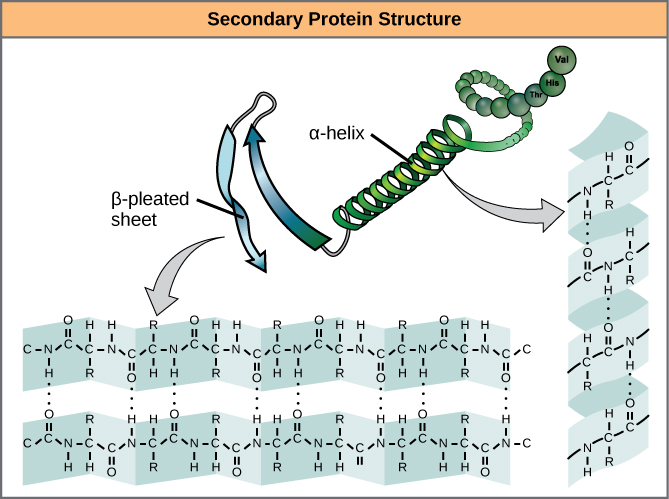 alpha-helix and beta-sheet ornaments in proteins. The hydrogen alliances formation as the protein is being expressed. Source: OpenStax Biology
alpha-helix and beta-sheet ornaments in proteins. The hydrogen alliances formation as the protein is being expressed. Source: OpenStax Biology
Proteins combine these two motifs, as well as variants on their topics, to form the next grade of organization, the tertiary arrangement. Unlike the simple themes of the secondary structure, tertiary organization tends to be driven more by hydrophobicity. Most proteins tend to have highly hydrophobic amino battery-acids, like alanine and methionine, at their core, where water is eliminated due to the ” greasy ” mood of the residues. These designs will often show up in transmembrane proteins, which are embedded in the lipid bilayer membrane circumventing cadres. The hydrophobic domains on the protein are thermodynamically stable inside the fatty interior of the tissue, while the hydrophilic regions of the protein are exposed to the aqueous situation on the sides of the membrane.
Tertiary organizations also tend to be stabilized by long-distance alliances between amino battery-acids. The classic sample of this is the disulfide bridge, which is frequently appears between two cysteine residues. If you’ve ever been to a hair salon and smelled the modest rotten-egg stink of someone getting a perm, you’re witnessing the part denaturation of the tertiary arrangement of keratin in whisker by the reduction of disulfide bails squandering sulfur-containing thiol compounds.
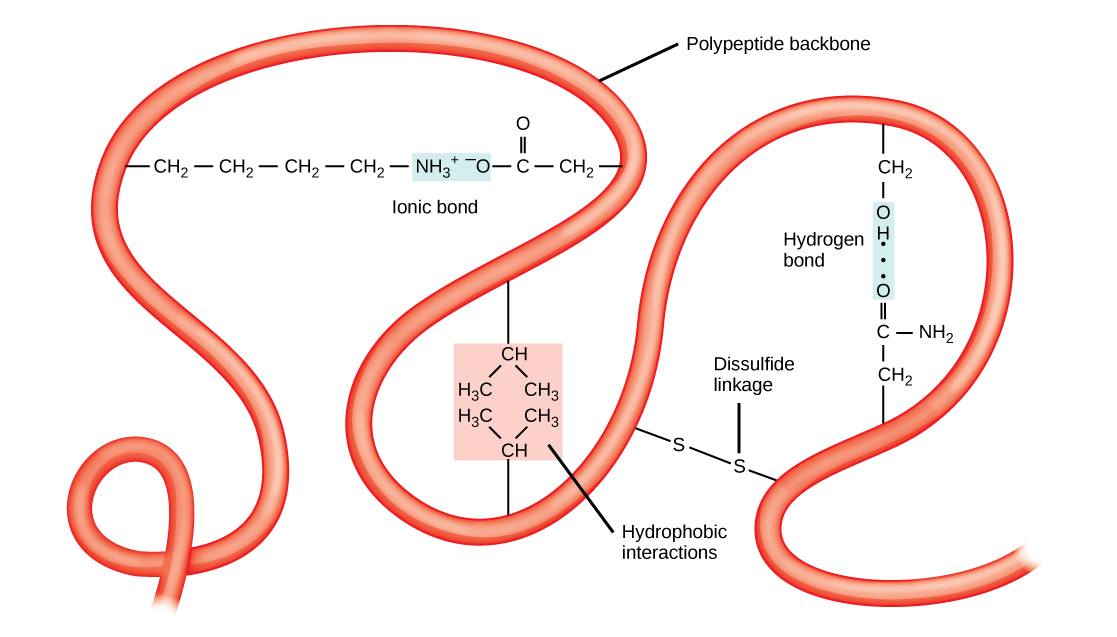 Tertiary structure is stabilized by long-distance interactions, like hydrophobicity and disulfide attachments. Source: OpenStax Biology
Tertiary structure is stabilized by long-distance interactions, like hydrophobicity and disulfide attachments. Source: OpenStax Biology
Disulfide connections can occur between cysteine residues in the same chain of polypeptides, or between cysteines set in accomplished different bonds. Interactions between different polypeptide bonds are the fourth height of protein arrangement, the quaternary organization. The haemoglobin in your blood is a perfect example of quaternary arrangement. Each hemoglobin molecule is constituted by four indistinguishable globin protein subunits, each of which is held in a specific conformation by disulfide connects within the polypeptide as well as ligament with the iron-containing heme molecule. All four globin subunits are to be bound by intermolecular disulfide connects, and the entire molecule acts as one to bind up to four oxygen molecules at once, and to liberate them when needed.
Simulate Organizations In a Search for Mixtures to the Illness
Polypeptide chains begin folding into their final shape during translation, as the growing chain is extruded from the ribosome, same to the way a piece of arranged remembering wire can snarl into a complex chassis when heated. But as is always the case with biology, there’s much more to the story.
In many cells, there is extensive editing of the rewritten genes that are available before translation, which reforms the primary design hugely compared to the raw base sequence of the gene. The translational system also often mobilizes the help of molecular chaperones, proteins that temporarily bind to the nascent polypeptide chain to prevent it from taking an intermediate structure that they are able to prevent it from taking its final shape.
All this is to say that predicting the final mold of a protein from the primary arrangement is not incidental. For decades, the only way to explore protein structure was with physical techniques like X-ray crystallography. It wasn’t until the late 1960 s that biophysical pharmacists started structure computational frameworks for protein folding, focused mainly on modeling the secondary structure of a protein. These methodologies and their successors take a vast amount of input data in addition to the primary arrangement cycle, such as tables of attachment tilts between amino battery-acids, directories of hydrophobicity, commission districts, and even conservation of structure and perform over evolutionary timescales to make a best guess at what a protein is going to look like.
Current computational methods used to secondary formation prediction, like those running on Folding @Home’ s network right now, run at about 80% accuracy, which is pretty good considering the complexity of the duty. The data generated by the folding prediction sits for proteins like the SARS-CoV-2 spike protein will be coupled with physical study data to come up with a conglomerate structure for the protein, and perhaps contribute us insights into how the virus obliges to the human angiotension converting enzyme-2( ACE-2) receptors that way the respiratory tract, which is its path into the body. If we can figure out the structure, we might be able to find stimulants to block binding and prevent infection.
Protein folding research is central to our understanding of so many diseases and illness that even once we figure out a room to beat COVID-1 9, the Folding @Home structure, which as realized such explosive swelling over the past month, will not run idle for long. The network is a research tool well-suited to exploring protein mannequins central to dozens of ailments that are linked to misfolded proteins, such as Alzheimer’s and variant Cruetzfeldt-Jakob disease, often incorrectly called mad-cow disease. And when the next virus surely comes along, all that horsepower, and all the experience being gained in managing it, will be ready to go again.
Read more: hackaday.com




Recent Comments